Talasseemia määratlus
Talasseemia on geneetiliselt leviv verehaigus, mille puhul keha sünteesib ebanormaalset hemoglobiini.
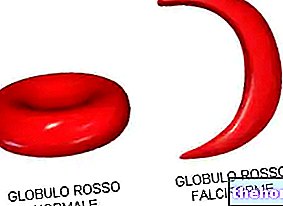
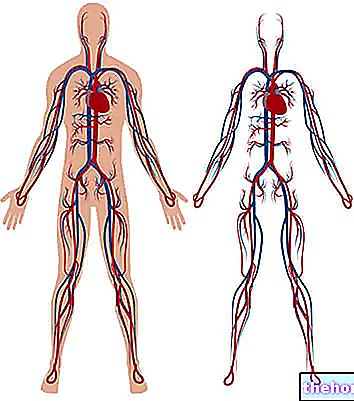
Nagu enamik inimesi teab, on hemoglobiin punaste vereliblede valk, mis on oluline hapniku transportimiseks veres. Talasseemia all kannatavatel isikutel põhjustab muteerunud hemoglobiini vorm punaste vereliblede järkjärgulist, kuid vääramatut hävitamist kuni aneemia tekkeni.
Meditsiinilise statistika põhjal on selge, et talasseemia mõjutab peamiselt Lähis -Ida riikide, Aafrika riikide elanikke ja kõiki neid, kes asustavad soiseid kohti (pole üllatav, et ka talasseemiat nimetatakse Vahemere aneemia).
Klassifikatsioon ja põhjused
Defektse valgu allüksuse (mis moodustab hemoglobiini) järgi eristatakse kahte talasseemia vormi; enne analüüsi jätkamist astume sammu tagasi, et selgitada mõningaid väga olulisi mõisteid.
Hemoglobiin on par excellence kandja, mida kasutatakse hapniku transportimiseks veres; see koosneb kahest valgust, mida tuntakse alfa-globuliini ja beeta-globuliini nime all.
Talasseemia tekib siis, kui üks või mitu ühe või mõlema valgu tootmist kontrollivat geeni on defektsed (muteerunud).
Talasseemia on põhjustatud hemoglobiini moodustavate valkude DNA mutatsioonist: need muutused mõjutavad tugevalt hemoglobiini füsioloogilist sünteesi ja põhjustavad erütrotsüütide hävitamist aneemia.
Talasseemia klassifitseerimisel tuleb lähtuda kahest olulisest tegurist:
- Vanematelt päritud muteerunud geenide arv
- Kaasatud valgu tüüp (alfa- või beeta -hemoglobiin)
Alfa -talasseemia
Talasseemia "alfa" vormis - kui hemoglobiini 4 "alfa" kerakujulist alaühikut (kromosoomis 16) saab muteerida - on tegemist ühe või mitme defektse geeniga; iga kerajas alaühik on selgelt kodeeritud geenist, seega kaasatavad geenid on 4.

Üldine sümptomipilt muutub tõsisemaks, kui on kaasatud kolm või neli geeni: esimesel juhul räägime "Hemoglobiini H haigus"(Mõõdukate või raskete sümptomitega). Kui kaasatud on kõik neli geeni, nimetatakse haigust suur alfa-talasseemia: sarnastes olukordades sureb vastsündinu vahetult enne sündi või varsti pärast seda.
Beeta -talasseemia
Talasseemia beeta vorm, nagu võib arvata, tekib siis, kui beetahelate koostises osalevad geenid on muteerunud (kromosoomi 11 tasemel): sel juhul saab mõjutada ainult kahte geeni. Kui muudetakse ainult ühte geeni, nimetatakse seda väike beeta-talasseemia, mille puhul patsient ei kaeba olulisi sümptomeid. Sarnaselt alfa -variandiga annab mõlema geeni kaasamine hemoglobiini beeta -ahelaid ühe suur beeta-talasseemia (või Cooley aneemia), mis peegeldab tõsiseid ja raskeid sümptomeid; sel juhul aga algavad sümptomid tavaliselt paari aasta pärast sünnist.
Vaata videot
- Vaata videot youtubest
Sümptomid
Lisateabe saamiseks: Talasseemia sümptomid
Talasseemia on väga tõsine pärilik haigus, nii et mõned selle variandid, näiteks suur alfa-talasseemia, võivad põhjustada lapse surma sünnituse ajal või varsti pärast sündi. Beeta-talasseemiaga väikelapsed võivad siiski ellu jääda ja areneda esimesed sümptomid paari aasta jooksul pärast sündi (raske aneemia).
Kui talassemia alfa- ja beetavormis on muudetud ainult ühte geeni, ei kurda patsiendid märgatavate sümptomite üle; ainult patsiendilt võetud vereproovi mikroskoobi analüüsi abil on erütrotsüütide kuju ja struktuuri kõrvalekalle normist palju väiksem.
Lisaks aneemiale võivad talasseemiaga patsiendid kogeda ühte või mitut järgmistest sümptomitest: väsimus, meeleolu muutused (ärrituvus), kasvuhäired, näo luude deformatsioonid, ikterus, õhupuudus ja tume uriin.
Raskuste korral võib talasseemia all kannatava patsiendi sümptomaatiline pilt taandareneda, luues tõelisi luude deformatsioone, eriti näol ja koljus; talasseemia võib soodustada "luuüdi ebanormaalset laienemist, muutes luumassi habras ja suurendades tohutult luumurdude riski.
Talasseemia tüsistuste hulgas tuleks mainida ka raua võimalikku kogunemist (hemokromatoos), mis väljendub nii haiguses kui ka korduvates vereülekannetes, mida patsient vajab.
Talasseemia põhjustab sageli splenomegaaliat, see tähendab põrna liialdatud mahulist suurenemist: sageli nõuab see patoloogiline kliiniline seisund splenektoomiat, elundi kirurgilist eemaldamist. Nagu me teame, on põrn oluline organ, mida kasutatakse vererakkude sünteesiks ja antikehad, lisaks infektsioonide tõrjele: selle eemaldamine soodustab selgelt kaitsefunktsiooni vähenemist bakteriaalsete ja viiruslike solvangute eest, muutes katsealuse infektsioonide suhtes tundlikumaks. Siiski tuleb märkida, et ka talasseemia suurendab riski haigestuda. nakatumiseks: põrna ekstsisiooni korral talasseemia taustal suureneb nakatumise võimalus liialdatult.

Diagnoos
Kui isa ja / või ema kannatab talasseemia all, on haiguse edasikandumise tõenäosus järglastele väga suur.Oleme analüüsinud, et mitte kõik talasseemia vormid ei alga täpse sümptomaatikaga kohe sünnist alates: sarnastes olukordades on talasseemia kahtluse korral võimalik patsiendile teha mitmeid spetsiifilisi teste ja uuringuid, mille eesmärk on diagnoosida ( (nt beeta-talasseemilisi geene kandvatel tervetel isikutel kõrgenenud hemoglobiin A2 määramine).
Füüsiliste uuringute hulgas võib põrna meditsiiniline palpatsioon mõnikord kindlaks teha talasseemia: splenomegaalia, nagu varem mainitud, on esimene häiresignaal Vahemere aneemia kohta. Vereanalüüsid on täpsemad ja täpsemad: talasseemia vereproovis tunduvad punased verelibled mikroskoobi all vaadatuna väikesed ja ebanormaalse kujuga. Lisaks näitab talasseemiat põdeva patsiendi hoolikas vereanalüüs tõsist aneemiat: see test on kasulik vere rauasisalduse määramiseks, DNA -analüüsi tegemiseks haiguse diagnostiliseks hindamiseks ja hemoglobiini võimaliku mutatsiooni hindamiseks. .
Teisest küljest näitab hemoglobiinide elektroforees hapnikku kandvate valkude ebanormaalset kuju.
Mõnda talasseemia varianti ei saa elektroforeesiga diagnoosida: sel juhul tehakse patsiendile "mutatsioonianalüüsi" test, mis on kasulik talasseemia avastamiseks ja kindlakstegemiseks.
Ravimid ja ravi
Vaata ka: Ravimid talasseemia raviks
Kuna tegemist on geneetiliselt leviva haigusega, on mõistetav, et praegu pole ühtegi ravimit, mis suudaks haigust tagasi pöörata; siiski on võimalik sümptomeid kontrollida, parandades patsiendi elukvaliteeti. Ühe või teise ravi valik sõltub talasseemia tüübist ja sümptomite tõsidusest.
Talasseemia kerge variandi korral (mille puhul muudetakse näiteks ainult ühte geeni) pole ravimeid vaja, kuna patsient ei kurda sümptomite üle. Sellistes oludes on siiski soovitatav regulaarselt vajalikke kontrolle läbi viia; Mõnikord on mõnikord kasulikud aeg -ajalt vereülekanded (eriti operatsiooni ja sünnituse korral).
Mõõduka või raske sümptomaatilise vormi korral on raviviis erinev ja võib nõuda sagedast vereülekannet või rasketel juhtudel tüvirakkude siirdamist.
- Vereülekanne: see terapeutiline lähenemisviis võib tekitada ka tõsiseid tüsistusi, kuna sagedased vereülekanded võivad soodustada raua patoloogilist kogunemist veres (hemokromatoos), mis nõuab spetsiifilist ravi, mille eesmärk on kõrvaldada raua säilitamine, mida nimetatakse teraapia kelaatoriks (selliste ravimitega nagu Deferasirox ja Deferiproon). Lisateabe saamiseks lugege artiklit hemokromatoosi raviks kasutatavate ravimite kohta.
- Luuüdi siirdamine: reserveeritud kõige tõsisematele juhtumitele, mille puhul talasseemia tekitab kehas tõsiseid häireid.
.jpg)




-ricotta-facile-e-veloce.jpg)