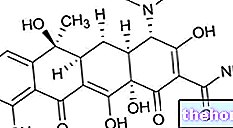
Toimeained: lamivudiin
Zeffix 5 mg / ml suukaudne lahus
Zeffixi pakendi infolehed on saadaval järgmistes pakendisuurustes:- Zeffix 100 mg õhukese polümeerikattega tabletid
- Zeffix 5 mg / ml suukaudne lahus
Miks Zeffixit kasutatakse? Milleks see mõeldud on?
Zeffixi toimeaine on lamivudiin.
Zeffixi kasutatakse kroonilise (pikaajalise) B-hepatiidi infektsiooni raviks täiskasvanutel.
Zeffix on viirusevastane ravim, mis pärsib B -hepatiidi viirust ja kuulub ravimite rühma, mida nimetatakse nukleosiidi analoogi pöördtranskriptaasi inhibiitoriteks (NRTI).
B-hepatiidi põhjustab viirus, mis nakatab maksa, põhjustab kroonilist (pikaajalist) infektsiooni ja võib maksa kahjustada. Zeffixi võib kasutada inimestel, kellel on maksakahjustus, kuid mis toimib endiselt normaalselt (kompenseeritud maksahaigus), ja kombinatsioonis teiste ravimitega inimestel, kellel on maksakahjustus ja mis ei tööta normaalselt (dekompenseeritud maksahaigus).
Ravi Zeffixiga võib vähendada B -hepatiidi viiruse hulka organismis. See peaks kaasa tooma maksakahjustuse vähenemise ja maksafunktsiooni paranemise. Mitte kõik inimesed ei reageeri Zeffix -ravile ühtemoodi. Arst kontrollib ravi efektiivsust regulaarsete vereanalüüsidega.
Vastunäidustused Zeffixi ei tohi kasutada
Ärge võtke Zeffixi
- kui olete lamivudiini või selle ravimi mis tahes koostisosa (de) suhtes allergiline
- Rääkige oma arstiga, kui arvate, et see kehtib teie kohta.
Ettevaatusabinõud kasutamisel Mida on vaja teada enne Zeffixi võtmist
Mõnedel Zeffixi või muid sarnaseid ravimeid kasutavatel inimestel on suurem tõsiste kõrvaltoimete oht. Peate olema teadlik järgmistest lisariskidest:
- kui teil on kunagi olnud muud tüüpi maksahaigusi nagu C -hepatiit
- kui teil on tõsine ülekaal (eriti kui olete naine).
- Rääkige oma arstile, kui mõni neist kehtib teie kohta. Ravimi võtmise ajal võib osutuda vajalikuks täiendav kontroll, sealhulgas vereanalüüsid.
Ärge lõpetage Zeffixi võtmist ilma arsti ettekirjutuseta, kuna on hepatiidi ägenemise oht. Kui te lõpetate Zeffixi võtmise, jälgib arst teid vähemalt neli kuud, et kontrollida, kas teil on probleeme. See hõlmab vereproovide võtmist, et kontrollida maksaensüümide taseme tõusu, mis võib viidata maksakahjustusele. Lisateavet Zeffixi võtmise kohta leiate lõigust 3.
Teiste inimeste kaitse
B -hepatiidi nakkus levib seksuaalvahekorras nakatunud inimestega või nakatunud vere ülekande kaudu (näiteks süstlanõelte vahetamise kaudu). Zeffix ei takista B -hepatiidi nakkuse levikut teistele inimestele. Et kaitsta teisi inimesi B -hepatiidi nakkuse eest:
- kasutage oraalseks või läbitungivaks seksiks kondoomi.
- ärge riskige kokkupuudet verega - näiteks ärge vahetage nõelu.
Koostoimed Millised ravimid või toiduained võivad Zeffixi toimet muuta
Teatage oma arstile või apteekrile, kui te kasutate, olete hiljuti kasutanud või kavatsete kasutada mis tahes muid ravimeid, kaasa arvatud taimseid ravimeid või muid ilma retseptita ostetud ravimeid.
Ärge unustage oma arstile või apteekrile öelda, kui võtate Zeffixi võtmise ajal uusi ravimeid.
Neid ravimeid ei tohi koos Zeffixiga võtta:
- muud lamivudiini sisaldavad ravimid, mida kasutatakse HIV -nakkuse raviks (mõnikord nimetatakse seda ka AIDS -i viiruseks)
- emtritsitabiini, mida kasutatakse HIV -nakkuse või B -hepatiidi viirusnakkuse raviks
- kladribiin, mida kasutatakse karvrakulise leukeemia raviks
- Rääkige oma arstile, kui teid ravitakse mõne nimetatud ravimiga.
Hoiatused Oluline on teada, et:
Rasedus
Kui olete rase, kahtlustate või kavatsete rasestuda:
- arutage oma arstiga Zeffixi võtmise riske ja kasu raseduse ajal. Ärge lõpetage Zeffixi võtmist ilma arsti nõuanneteta.
Toitmisaeg
Zeffix võib imenduda rinnapiima. Kui te toidate last rinnaga või kavatsete seda teha:
- enne Zeffixi võtmist pidage nõu oma arstiga.
Autojuhtimine ja masinatega töötamine
Zeffix võib põhjustada väsimustunnet, mis võib mõjutada teie autojuhtimise ja masinate käsitsemise võimet.
- Ärge juhtige autot ega töötage masinatega, kui tunnete end väsinuna.
Zeffix sisaldab suhkrut ja säilitusaineid
Kui teil on diabeet, pidage meeles, et iga Zeffixi annus (100 mg = 20 ml) sisaldab 4 g sahharoosi.
Zeffix sisaldab sahharoosi. Kui arst on teile öelnud, et te ei talu teatud suhkruid, võtke enne Zeffixi võtmist ühendust oma arstiga. Sahharoos võib teie hambaid kahjustada.
Zeffix sisaldab ka säilitusaineid (parahüdroksübensoaate), mis võivad põhjustada allergilisi reaktsioone (mis võivad tekkida ka viivitusega).
Annus, manustamisviis ja aeg Kuidas Zeffixit kasutada: Annustamine
Võtke seda ravimit alati täpselt nii, nagu arst on teile rääkinud. Kahtluse korral pidage nõu oma arsti või apteekriga.
Püsige pidevas kontaktis oma arstiga
Zeffix aitab kontrollida B -hepatiidi infektsiooni. Infektsiooni tõrjeks ja haiguse süvenemise vältimiseks peate seda võtma iga päev.
- Pidage ühendust oma arstiga ja ärge lõpetage Zeffixi võtmist ilma arsti nõuanneteta.
Kogus võtta
Tavaline Zeffixi annus on 20 ml (100 mg lamivudiini) üks kord ööpäevas.
Arst võib teile määrata väiksema annuse, kui teil on probleeme neerudega. Zeffixi suukaudne lahus on saadaval inimestele, kes vajavad soovitatust väiksemat annust või kes ei saa tablette võtta.
- Rääkige oma arstiga, kui see kehtib teie kohta.
Kui te võtate juba mõnda teist HIV -nakkuse raviks lamivudiini sisaldavat ravimit, jätkab arst teie ravi suurema annusega (tavaliselt 150 mg kaks korda ööpäevas), kuna Zeffixi lamivudiini annusest (100 mg) ei piisa. HIV -nakkus. Kui kavatsete oma HIV -ravi muuta, rääkige sellest kõigepealt oma arstiga.
Neelake tablett alla tervelt koos veega. Zeffixi võib võtta koos toiduga või ilma.
Kui te unustate Zeffixi võtta
Kui te unustate annuse võtmata, võtke see niipea, kui see teile meenub. Seejärel jätkake ravi nagu varem. Ärge võtke kahekordset annust, kui annus jäi eelmisel korral võtmata.
Ärge lõpetage Zeffixi võtmist
Ärge lõpetage Zeffixi võtmist ilma arstiga nõu pidamata. Hepatiidi ägenemise oht on ohtlik (vt lõik 2). Kui te lõpetate Zeffixi võtmise, jälgib arst teid vähemalt neli kuud, et kontrollida probleeme. See hõlmab vereproovide võtmist, et kontrollida maksaensüümide taseme tõusu, mis võib viidata maksakahjustusele.
Üleannustamine Mida teha, kui olete võtnud liiga palju Zeffixi
Zeffixi liiga palju kogemata võtmine ei põhjusta tõenäoliselt tõsiseid probleeme. Kui te võtate kogemata liiga palju, rääkige sellest oma arstile või apteekrile või võtke ühendust lähima haigla erakorralise meditsiini osakonnaga.
Kõrvaltoimed Millised on Zeffixi kõrvaltoimed
Nagu kõik ravimid, võib ka see ravim põhjustada kõrvaltoimeid, kuigi kõigil neid ei teki.
Zeffixi kliinilistes uuringutes sageli teatatud kõrvaltoimed olid väsimus, hingamisteede infektsioonid, kurguvalu, peavalu, mao- ja kõhuvalu, iiveldus, oksendamine ja kõhulahtisus, maksaensüümide ja ensüümide sisalduse suurenemine lihastes (vt allpool).
Allergiline reaktsioon
Need on haruldased (võivad esineda kuni 1 inimesel 1000 -st). Märgid hõlmavad järgmist:
- silmalaugude, näo või huulte turse
- neelamis- või hingamisraskused.
- Nende sümptomite ilmnemisel võtke kohe ühendust oma arstiga. Lõpetage Zeffixi võtmine.
Arvatavasti põhjustas Zeffix kõrvaltoimeid
Väga sageli esinevad kõrvaltoimed (võivad esineda rohkem kui 1 inimesel 10 -st), mis võivad ilmneda vereanalüüsides, on:
- teatud maksaensüümide (transaminaaside) taseme tõus, mis võib olla märk põletikust või maksakahjustusest.
Sagedased kõrvaltoimed (võivad esineda kuni 1 inimesel 10 -st) on:
- krambid ja lihasvalu
- lööve või nõgestõbi kõikjal kehal
Sagedased kõrvaltoimed, mis võivad ilmneda vereanalüüsides, on järgmised:
- lihastes toodetud ensüümi (kreatiinfosfokinaas) taseme tõus, mis võib olla märk koekahjustusest.
Väga harv kõrvaltoime (võib esineda kuni 1 inimesel 10 000 -st) on:
- piimatsidoos (piimhappe liig veres).
Muud kõrvaltoimed
Teisi kõrvaltoimeid on esinenud väga vähesel arvul inimestel, kuid nende täpne esinemissagedus ei ole teada:
- lihaskoe lagunemine
- maksahaiguse süvenemine pärast Zeffixi kasutamise lõpetamist või ravi ajal, kui B -hepatiidi viirus muutub Zeffixi suhtes resistentseks. See võib mõnedel inimestel lõppeda surmaga.
Kõrvaltoimed, mis võivad ilmneda vereanalüüsides, on järgmised:
- vere hüübimisega seotud rakkude arvu vähenemine (trombotsütopeenia).
Kui teil tekivad kõrvaltoimed
- Rääkige sellest oma arstile või apteekrile. See hõlmab ka kõiki võimalikke kõrvaltoimeid, mida selles infolehes ei ole nimetatud.
Kõrvaltoimetest teatamine
Kui teil tekib ükskõik milline kõrvaltoime, pidage nõu oma arsti või apteekriga, sealhulgas selles infolehes loetlemata. Te võite ka teatada kõrvaltoimetest otse riikliku teavitussüsteemi kaudu. Kõrvaltoimetest teatades saate aidata saada lisateavet selle ravimi ohutuse kohta.
Aegumine ja säilitamine
Hoidke seda ravimit laste eest varjatud ja kättesaamatus kohas.
Ärge kasutage seda ravimit pärast kõlblikkusaega, mis on märgitud karbil ja blistril.
Hoida temperatuuril kuni 30 ° C.
Ärge visake ravimeid kanalisatsiooni ega olmejäätmete hulka. Küsige oma apteekrilt, kuidas visata ära ravimeid, mida te enam ei kasuta. See aitab kaitsta keskkonda.
Muu info
Mida Zeffix sisaldab
Toimeaine on lamivudiin. Üks õhukese polümeerikattega tablett sisaldab 100 mg lamivudiini.
Abiained on: mikrokristalne tselluloos, naatriumtärklisglükolaat, magneesiumstearaat, hüpromelloos, titaandioksiid, makrogool 400, polüsorbaat 80, sünteetilised kollased ja punased raudoksiidid.
Kuidas Zeffix välja näeb ja pakendi sisu
Zeffix õhukese polümeerikattega tabletid on turvakarpides, mis sisaldavad 28 või 84 tabletti sisaldavaid alumiiniumblistreid.
Tabletid on karamellivärvi, õhukese polümeerikattega, kapslikujulised, kaksikkumerad, ühele küljele on pressitud "GX CG5".
Kõik pakendi suurused ei pruugi olla müügil.
Allika pakendi infoleht: AIFA (Itaalia ravimiamet). Sisu avaldati jaanuaris 2016. Esitatud teave ei pruugi olla ajakohane.
Kõige ajakohasemale versioonile juurdepääsu saamiseks on soovitatav külastada AIFA (Itaalia ravimiamet) veebisaiti. Vastutusest loobumine ja kasulik teave.
01.0 RAVIMPREPARAADI NIMETUS
ZEFFIX 5 MG / ML SUULINE LAHUS
02.0 KVALITATIIVNE JA KVANTITATIIVNE KOOSTIS
Üks ml suukaudset lahust sisaldab 5 mg lamivudiini
Teadaolevat toimet omavad abiained:
20% sahharoos (4 g / 20 ml)
Metüülparahüdroksübensoaat (E218) 1,5 mg / ml
Propüülparahüdroksübensoaat (E216) 0,18 mg / ml
Abiainete täielik loetelu vt lõik 6.1.
03.0 RAVIMVORM
Suukaudne lahus.
Selge, värvitu kuni kahvatukollane.
04.0 KLIINILINE TEAVE
04.1 Näidustused
Zeffix on näidustatud kroonilise B -hepatiidi raviks täiskasvanud patsientidel, kellel on:
• kompenseeritud maksahaigus, millel on tõendeid aktiivse viiruse replikatsiooni kohta, pidevalt suurenenud seerumi alaniinaminotransferaasi (ALT) tase ja histoloogilised tõendid aktiivse maksapõletiku ja / või fibroosi kohta. Lamivudiinravi alustamist tuleks kaaluda ainult siis, kui alternatiivse viirusevastase aine kasutamine, millel on suurem geneetiline resistentsusbarjäär, ei ole kättesaadav või asjakohane (vt lõik 5.1).
• dekompenseeritud maksahaigus kombinatsioonis teise ravimiga, millel puudub ristresistentsus lamivudiini suhtes (vt lõik 4.2).
04.2 Annustamine ja manustamisviis
Annustamine
Zeffix -ravi peab alustama kroonilise B -hepatiidi ravis kogenud arst.
Täiskasvanud: Zeffixi soovitatav annus on 100 mg üks kord ööpäevas.
Dekompenseeritud maksahaigusega patsientidel tuleb lamivudiini alati kasutada kombinatsioonis teise viirusevastase ainega, millel puudub ristresistentsus lamivudiini suhtes, et vähendada resistentsuse ohtu ja saavutada kiire viiruse supressioon.
Ravi kestus: Ravi optimaalne kestus ei ole teada.
• patsientidel, kellel on tsirroosita HBeAg-positiivne krooniline B-hepatiit (CHB), tuleb ravi manustada vähemalt 6-12 kuud pärast HBeAg serokonversiooni (HBeAg ja HBV DNA kadumine koos HBeAb tuvastamisega) kinnitamist, et vähendada riski viroloogiline retsidiiv või kuni HBsAg serokonversiooni või efektiivsuse kadumiseni (vt lõik 4.4). Pärast ravi lõpetamist tuleb regulaarselt jälgida seerumi ALAT- ja HBV -DNA taset, et avastada hilinenud viroloogiline retsidiiv.
• patsientidel, kellel on HBeAg negatiivne CHB (tuumorieelsed mutandid) ilma tsirroosita, tuleb ravi läbi viia vähemalt kuni HBs-i serokonversioonini või kui on tõendeid efektiivsuse vähenemise kohta. Pikaajalise ravi korral on soovitatav regulaarselt jälgida, et veenduda, et valitud ravi jätkamine on patsiendile sobiv.
• ravi katkestamist ei soovitata dekompenseeritud maksahaiguse või tsirroosiga patsientidel ega maksa siirdamisel (vt lõik 5.1).
Pärast Zeffix -ravi katkestamist tuleb patsiente perioodiliselt jälgida ägenemise hepatiidi suhtes (vt lõik 4.4).
Kliiniline resistentsus: CHB-ga patsientidel, nii HBeAg positiivne kui ka HBeAg negatiivne, võib HBV YMDD (türosiin-metioniin-aspartaat-aspartaat) mutandi areng põhjustada lamivudiini ravivastuse vähenemist, mida näitab HBV DNA ja ALAT suurenemine võrreldes ravi varasematele tasemetele. Resistentsuse ohu vähendamiseks lamivudiini monoteraapiaga ravitavatel patsientidel tuleb kaaluda ravi muutmist, kui HBV DNA jääb tuvastatavaks 24 või enama ravinädala jooksul. HBV YMDD mutandiga patsientidel tuleb seda kaaluda. -resistentsus lamivudiini suhtes (vt lõik 5.1).
Spetsiaalsed populatsioonid
Lapsed
Zeffixi ohutus ja efektiivsus lastel ja alla 18 -aastastel noorukitel ei ole tõestatud. Praegu kättesaadavad andmed on kirjeldatud lõikudes 4.4 ja 5.1, kuid annustamissoovitusi ei saa anda.
Neerupuudulikkus
Mõõduka kuni raske neerukahjustusega patsientidel suureneb lamivudiini kontsentratsioon seerumis (AUC) neerukliirensi vähenemise tõttu. Seetõttu tuleb annust vähendada patsientidel, kelle kreatiniini kliirens on alla 50 ml / minutis. Kui on vaja alla 100 mg annuseid, tuleb kasutada Zeffix suukaudset lahust (vt tabel 1 allpool).
Tabel 1: Zeffixi annus vähenenud renaalse kliirensiga patsientidel.
Olemasolevad andmed patsientide kohta, kes saavad vahelduvat hemodialüüsi (kestusega kuni 4 tundi dialüüsi 2-3 korda nädalas), näitavad, et pärast lamivudiini algannuse vähendamist kreatiniini kliirensi kompenseerimiseks ei ole dialüüsi ajal muud vaja muuta annust.
Maksapuudulikkus
Maksapuudulikkusega patsientide, sealhulgas siirdamist ootavate kaugelearenenud maksahaigusega patsientide kohta saadud andmed näitavad, et maksafunktsiooni kahjustus ei mõjuta oluliselt lamivudiini farmakokineetikat. Nende andmete põhjal ei ole maksapuudulikkusega patsientidel annuse kohandamine vajalik, välja arvatud juhul, kui sellega kaasneb neerupuudulikkus.
Manustamisviis
Zeffixi võib võtta koos toiduga või ilma.
04.3 Vastunäidustused
Ülitundlikkus toimeaine või lõigus 6.1 loetletud mis tahes abiainete suhtes.
04.4 Erihoiatused ja asjakohased ettevaatusabinõud kasutamisel
Lamivudiini on manustatud kompenseeritud kroonilise B -hepatiidiga lastele (2 -aastased ja vanemad) ja noorukitele. Andmete piiratuse tõttu ei soovitata selles patsiendipopulatsioonis lamivudiini manustamist praegu (vt lõik 5.1).
Lamivudiini efektiivsus samaaegse Delta -hepatiidi või C -hepatiidi infektsiooniga patsientidel ei ole tõestatud ja soovitatav on olla ettevaatlik.
Lamivudiini kasutamise kohta HBeAg-negatiivsetel (tuumorieelsetel mutantidel) patsientidel ja patsientidel, kes saavad samaaegselt immunosupressiivseid raviskeeme, sealhulgas vähi keemiaravi, on vähe andmeid. Sellistel patsientidel tuleb lamivudiini kasutada ettevaatusega.
Patsiente tuleb Zeffix -ravi ajal regulaarselt jälgida. Seerumi ALAT ja HBV DNA taset tuleb jälgida 3 -kuuliste intervallidega ning HBeAg -positiivsetel patsientidel tuleb HBeAg -i hinnata iga 6 kuu järel.
Hepatiidi ägenemine
Ägenemine ravi ajal: Kroonilise B -hepatiidi spontaansed ägenemised on suhteliselt tavalised ja neid iseloomustab seerumi ALAT -i mööduv tõus. Pärast viirusevastase ravi alustamist võib mõnedel patsientidel seerumi ALAT suureneda, samas kui HBV DNA tase seerumis väheneb. Kompenseeritud maksahaigusega patsientidel ei kaasnenud seerumi ALAT tõusuga üldiselt bilirubiini kontsentratsiooni suurenemist seerumis ega maksa dekompensatsiooni tunnuseid.
Pikaajalise ravi korral on tuvastatud HBV viiruse alampopulatsioonid, kellel on vähenenud tundlikkus lamivudiini (HBV YMDD mutant) suhtes. Mõnel patsiendil võib HBV YMDD mutandi areng põhjustada hepatiidi ägenemist, mida tõendavad peamiselt seerumi ALAT väärtuste tõus ja HBV taastumine DNA (vt lõik 4.2). Patsientidel, kellel on HBV YMDD mutant, tuleb kaaluda teise, rist-resistentsuseta aine lisamist lamivudiini suhtes (vt lõik 5.1).
Ägenemine pärast ravi lõpetamist: patsientidel, kes olid katkestanud B-hepatiidi ravi, täheldati ägedat hepatiidi ägenemist ja seda tõestas üldiselt seerumi ALAT taseme tõus ja HBV-DNA taasilmumine. Kontrollitud III faasi uuringutes, kus ei olnud aktiivset järelravi, oli ALAT taseme tõus pärast ravi (rohkem kui kolm korda algväärtusest) lamivudiiniga ravitud patsientidel (21%) suurem kui platseebot saanud patsientidel (8%). patsientide protsent, kellel oli ravijärgne bilirubiinisisalduse tõus, madalam ja sarnane mõlemas ravirühmas. Lisateavet ravi järgsete ALAT-i suurenemise sageduse kohta vt lõigust 5.1 tabelis 3. Lamivudiiniga ravitud patsientide puhul enamus ravi järgsetest ALAT tõusudest tekkis 8… 12 nädalat pärast ravi. Enamik juhtudest olid iseenesest piiravad, kuid siiski esinesid. täheldati mõningaid surmajuhtumeid. . t maksafunktsioon seerumis (ALAT ja bilirubiini tase) vähemalt neli kuud ja seejärel vastavalt kliinilisele praktikale.
Dekompenseeritud tsirroosiga patsientide ägenemine: Siirdatud retsipientidel ja dekompenseeritud tsirroosiga patsientidel on suurem oht aktiivseks viiruse replikatsiooniks. Kuna neil patsientidel on maksafunktsiooni kahjustus, võib hepatiidi taasaktiveerumine lamivudiini kasutamise katkestamise või efektiivsuse vähenemise tõttu ravi ajal põhjustada tõsist, isegi surmaga lõppevat dekompensatsiooni. Neid patsiente tuleb jälgida B -hepatiidiga seotud kliiniliste, viroloogiliste ja seroloogiliste näitajate suhtes, neeru- ja maksatalitluse ning viirusevastase toime suhtes ravi ajal (vähemalt iga kuu) ja kui ravi mingil põhjusel katkestatakse, siis vähemalt 6 kuud pärast ravi. Jälgitavad laboratoorsed parameetrid peaksid sisaldama (vähemalt) seerumi ALAT, bilirubiini, albumiini, BUN, kreatiniini ja viroloogilist seisundit: HBV antigeene / antikehi ja võimaluse korral HBV seerumi DNA kontsentratsioone. Patsiente, kellel ilmnevad maksapuudulikkuse nähud ravi ajal või pärast seda, tuleb vajaduse korral sagedamini jälgida.
Patsientide kohta, kellel on pärast ravi korduv hepatiit, ei ole piisavalt andmeid lamivudiini taasalustamise kasulikkuse kohta.
HIV kaasinfektsioon
HIV-nakkusega patsientidel, kes saavad või hakkavad saama lamivudiinravi või lamivudiini / zidovudiini kombinatsiooni, tuleb HIV-nakkuse jaoks ette nähtud lamivudiini annust (tavaliselt 150 mg kaks korda ööpäevas) säilitada päev koos teiste retroviirusevastaste ravimitega). HIV-nakkusega patsientidel, kes ei vaja retroviirusevastast ravi, on lamivudiini monoteraapia kroonilise B-hepatiidi raviks kasutamisel HIV-mutatsiooni oht.
B -hepatiidi edasikandumine
Puudub teave lamivudiiniga ravitud rasedate B-hepatiidi viiruse edasikandumise kohta emalt lootele. Tuleb järgida tavalisi B-hepatiidi viiruse vastu immuniseerimiseks soovitatud protseduure.
Patsiente tuleb teavitada, et lamivudiinravi ei ole näidanud, et see vähendaks hepatiit B viiruse ülekandumise riski. Seetõttu tuleb jätkata piisavate ettevaatusabinõude rakendamist.
Abiainete talumatus
Patsiendid, kellel on harvaesinev pärilik fruktoositalumatus, glükoosi-galaktoosi imendumishäire või sahharaasi-isomaltaasi puudulikkus, ei tohi seda ravimit kasutada.
Diabeediga patsient peab meeles pidama, et iga suukaudse lahuse annus (100 mg = 20 ml) sisaldab 4 g sahharoosi.
Suukaudne lahus sisaldab propüül- ja metüülparahüdroksübensoaati. Need ained võivad mõnedel inimestel põhjustada allergilist reaktsiooni. Seda reaktsiooni saab edasi lükata.
Laktatsidoos ja raske hepatomegaalia koos steatoosiga
Nukleosiidi analoogide kasutamisel on teatatud laktatsidoosi juhtudest (hüpokseemia puudumisel), mis võivad mõnikord lõppeda surmaga, tavaliselt raske hepatomegaalia ja maksa steatoosiga. Kuna Zeffix on nukleosiidi analoog, ei saa seda riski välistada. Nukleosiidi analoogide kasutamine tuleb lõpetada, kui aminotransferaaside tase tõuseb kiiresti, progresseeruv hepatomegaalia või teadmata etioloogiaga metaboolne / laktatsidoos. Seedetrakti mõjutavad mitte-tõsised sümptomid, nagu iiveldus, oksendamine ja kõhuvalu, võivad viidata laktatsidoosi arengule . Tõsiseid juhtumeid, mõnikord surmaga lõppenud, on seostatud pankreatiidiga, maksapuudulikkuse / rasvmaksaga, neerupuudulikkusega ja suurenenud seerumi laktaadisisaldusega. Ettevaatlik tuleb olla nukleosiidi analoogide määramisel patsientidele (eriti rasvunud naistele), kellel on hepatomegaalia, hepatiit või muud teadaolevad maksahaiguse ja rasvmaksahaiguse riskitegurid (sh mõned ravimid ja alkohol). Eriti ohtlikud võivad olla patsiendid, kes on samaaegselt nakatunud C -hepatiidiga ning keda ravitakse alfa -interferooni ja ribaviriiniga. Selliseid patsiente tuleb hoolikalt jälgida.
Mitokondriaalne düsfunktsioon
On näidatud, et nii nukleosiidi kui ka nukleotiidi analoogid on in vivo seda in vitro põhjustada erineval määral mitokondriaalseid kahjustusi. Nukleosiidi analoogidega kokku puutunud vastsündinutel on teatatud mitokondriaalse düsfunktsiooni juhtudest emakas ja / või pärast sündi. Peamised teatatud kõrvaltoimed on hematoloogilised häired (aneemia, neutropeenia), ainevahetushäired (hüperlaktateemia ja hüperlipaseemia). On teatatud hilinenud neuroloogilistest häiretest (hüpertoonia, krambid, käitumishäired). Neuroloogilised häired võivad olla mööduvad või püsivad. Iga laps paljastati emakas nukleosiidide ja nukleotiidide analoogide suhtes, tuleb läbi viia kliiniline ja laboratoorne järelkontroll ning neid tuleb hoolikalt jälgida võimalike mitokondrite düsfunktsioonide suhtes, kui ilmnevad seotud nähud ja sümptomid.
Zeffixi ei tohi võtta koos teiste lamivudiini või emtritsitabiini sisaldavate ravimitega.
Lamivudiini ja kladribiini kombinatsioon ei ole soovitatav (vt lõik 4.5).
04.5 Koostoimed teiste ravimitega ja muud koostoimed
Koostoimeuuringuid on läbi viidud ainult täiskasvanutel.
Metaboolsete koostoimete tõenäosus on väike, kuna ainevahetus on piiratud, plasmavalkudega vähe seondub ja aine muutumatul kujul peaaegu täielikult eritub neerude kaudu.
Lamivudiin elimineeritakse valdavalt aktiivse katioonsekretsiooni teel. Tuleb kaaluda koostoimete võimalust teiste samaaegselt manustatavate ravimitega, eriti kui nende esmane eliminatsioonitee on aktiivne sekretsioon neerude kaudu orgaanilise katioonide transpordisüsteemi, nt trimetoprimi abil. Teised ravimid (nt ranitidiin, tsimetidiin) elimineeritakse selle mehhanismi abil ainult osaliselt ja nende koostoime lamivudiiniga ei ole tõestatud.
Peamiselt aktiivse orgaanilise anioonisüsteemi või glomerulaarfiltratsiooni kaudu erituvad ained ei põhjusta kliiniliselt olulisi koostoimeid lamivudiiniga. 160 mg / 800 mg trimetoprimi / sulfametoksasooli manustamisel suureneb lamivudiini plasmakontsentratsioon ligikaudu 40%. Lamivudiin ei mõjuta trimetoprimi ega sulfametoksasooli farmakokineetikat. Lamivudiini annust ei ole vaja muuta, välja arvatud juhul, kui patsiendil on neerupuudulikkus.
Kombinatsioonis lamivudiiniga täheldati zidovudiini Cmax kerget tõusu (28%); üldine ekspositsioon (AUC) siiski oluliselt ei muutu Zidovudiin ei mõjuta lamivudiini farmakokineetikat (vt lõik 5.2).
Lamivudiinil ei ole nende ravimite samaaegsel manustamisel farmakokineetilisi koostoimeid alfa-interferooniga, kuid ametlikke koostoimeuuringuid ei ole läbi viidud.
Kladribiin: in vitro lamivudiin pärsib kladribiini rakusisest fosforüülimist, põhjustades kliinilises keskkonnas kombineerituna kladribiini efektiivsuse vähenemise riski.
Mõned tõendid toetavad ka lamivudiini ja kladribiini võimalikku koostoimet. Seetõttu ei ole lamivudiini ja kladribiini samaaegne manustamine soovitatav (vt lõik 4.4).
04.6 Rasedus ja imetamine
Rasedus
Suur hulk andmeid rasedate kohta (rohkem kui 1000 kokkupuutejuhtumit) ei näita väärarengutega seotud toksilisust. Zeffixi võib raseduse ajal kasutada, kui see on kliiniliselt vajalik.
Patsientidel, keda ravitakse lamivudiiniga ja seejärel rasestutakse, tuleb kaaluda hepatiidi kordumise võimalust pärast lamivudiini kasutamise lõpetamist.
Toitmisaeg
Võttes arvesse enam kui 130 ema-lapse paari, keda ravitakse HIV-iga, on lamivudiini kontsentratsioon seerumis rinnaga toidetavatel imikutel HIV-ravi saavatelt emadelt väga madal (ligikaudu 0,06–4% ema seerumi kontsentratsioonist) ja langeb rinnaga toitmise ajal järk-järgult tuvastamatutele tasemetele. imikud saavad 24 nädala vanuseks. Imetatava imiku neelatud lamivudiini üldkogus on väga väike ja seetõttu põhjustab see tõenäoliselt kokkupuuteid, millel on ebapiisav viirusevastane toime. Ema B -hepatiit ei põhjusta rinnaga toitmisele vastunäidustusi, kui imikut ravitakse adekvaatselt B -hepatiidi ennetamiseks sündides ja puuduvad tõendid selle kohta, et lamivudiini madal kontsentratsioon rinnapiimas põhjustab rinnaga toidetavatel imikutel soovimatuid toimeid. seda tuleb kaaluda imetavatel emadel, keda ravitakse lamivudiiniga HBV vastu, võttes arvesse rinnaga toitmise kasu lapsele ja ravi kasu emale. Kui HBV levib ema kaudu, vaatamata piisavale profülaktikale, tuleb kaaluda rinnaga toitmise katkestamist, et vähendada lamivudiini suhtes resistentsete mutantide tekkimise ohtu vastsündinul.
Viljakus
Andmed puuduvad.
Mitokondriaalne düsfunktsioon
On näidatud, et nii nukleosiidi kui ka nukleotiidi analoogid on in vivo seda in vitro põhjustada erineval määral mitokondriaalseid kahjustusi. Nukleosiidi analoogidega kokku puutunud vastsündinutel on teatatud mitokondriaalse düsfunktsiooni juhtudest emakas ja / või pärast sündi (vt lõik 4.4).
04.7 Mõju autojuhtimise ja masinate käsitsemise võimele
Ravimi toime kohta autojuhtimisele ja masinate käsitsemise võimele ei ole uuringuid läbi viidud.
04.8 Kõrvaltoimed
Kõrvaltoimete ja laboratoorsete kõrvalekallete esinemissagedus (välja arvatud ALAT ja CPK tõus, vt allpool) oli platseebo- ja lamivudiinravi saavatel patsientidel sarnane. Kõige sagedamini teatatud kõrvaltoimed olid halb enesetunne ja väsimus, hingamisteede infektsioonid, kurguvalu ja ebamugavustunne mandlites, peavalu, kõhuvalu või -krambid, iiveldus, oksendamine ja kõhulahtisus.
Kõrvaltoimed on loetletud allpool organsüsteemi klassi ja esinemissageduse järgi. Esinemissageduse kategooriad on määratud ainult nende kõrvaltoimete hulka, mida peetakse vähemalt põhjuslikult seotud lamivudiiniga. Esinemissagedused on määratletud järgmiselt: väga sage (≥ 1/10), sage (≥ 1/100 a
Kõrvaltoimete esinemissageduse kategooriad põhinevad peamiselt kliiniliste uuringute kogemustel, milles osales kokku 1171 kroonilise B -hepatiidi patsienti, keda raviti lamivudiiniga 100 mg.
* III faasi kliinilistes uuringutes lamivudiinirühmas täheldatud esinemissagedus ei olnud suurem kui platseeborühmas.
HIV-nakkusega patsientidel on teatatud pankreatiidi ja perifeersete neuropaatiate (või paresteesiate) juhtudest. Kroonilise B -hepatiidiga patsientidel ei täheldatud nende nähtude esinemissageduses lamivudiini ja platseeboga ravitud patsientide vahel erinevusi.
HIV -patsientidel kombineeritud ravi ajal nukleosiidi analoogidega on teatatud mõnikord surmaga lõppenud laktatsidoosi juhtudest, mis on tavaliselt seotud raske hepatomegaalia ja maksa steatoosiga.
Lamivudiiniga B -hepatiidi raviks ravitud patsientidel on teatatud harvadest laktatsidoosi juhtudest.
04.9 Üleannustamine
Lamivudiini manustamine eriti suurtes annustes ägeda toksilisuse loomkatsetes ei põhjustanud elunditoksilisust. Ägeda suukaudse üleannustamise tagajärgede kohta inimestel on vähe andmeid. Surmasid ei esinenud ja patsiendid paranesid. Üleannustamise järgselt ei tuvastatud mingeid spetsiifilisi tunnuseid ega sümptomeid.
Üleannustamise korral tuleb patsienti jälgida ja talle määrata sobiv standardne toetav ravi. Üleannustamise ravis võib kasutada pidevat hemodialüüsi, kuigi seda pole uuritud, kuna lamivudiin on dialüüsitav.
05.0 FARMAKOLOOGILISED OMADUSED
05.1 Farmakodünaamilised omadused
Farmakoterapeutiline rühm: viirusevastased ained süsteemseks kasutamiseks, nukleosiidid ja nukleotiidid pöördtranskriptaasi inhibiitorid.
ATC -kood: J05AF05.
Lamivudiin on viirusevastane aine, mis on aktiivne B -hepatiidi viiruse vastu kõigis testitud rakuliinides ja katseliselt nakatunud loomadel.
Nii tervetel kui ka nakatunud rakkudel metaboliseerub lamivudiin trifosfaadi derivaadiks (TP), mis on lähteaine aktiivne vorm. Trifosfaadi rakusisene poolväärtusaeg hepatotsüütides on 17–19 tundi in vitro. Lamivudiin-TP toimib viiruse HBV polümeraasi substraadina.
Edasise viiruse DNA moodustumist blokeerib lamivudiin-TP inkorporeerimine ahelasse ja sellele järgnev katkestamine.
Lamivudiin-TP ei häiri deoksünukleotiidide normaalset raku metabolismi. Samuti on see imetajate alfa- ja beeta -polümeraaside nõrk inhibiitor. Lisaks ei mõjuta lamivudiin-TP imetajarakkude DNA sisaldust vähe.
Analüüsides ainete võimalikku mõju mitokondriaalsele struktuurile ning DNA sisaldusele ja funktsioonile leiti, et lamivudiinil ei ole märgatavat toksilist toimet. Sellel on väga väike potentsiaal mitokondriaalse DNA sisalduse vähendamiseks, see ei ole mitokondriaalsesse DNA -sse püsivalt integreeritud ega toimi mitokondriaalse DNA polümeraasi gamma inhibiitorina.
Kliiniline kogemus
Kogemus HBeAg -positiivse CHB ja kompenseeritud maksahaigusega patsientidel: Kontrollitud uuringutes pärssis üks aasta lamivudiinravi oluliselt HBV DNA replikatsiooni [34–57% patsientidest olid alla testi avastamispiiride (Abbott Genostics lahuse hübridisatsioonitesti, LLOD pg / ml)], ALAT taseme normaliseerumine (40–72 % patsientidest), indutseeritud HBeAg serokonversioon (HBeAg kadumine ja HBeAb tuvastamine koos HBV DNA kadumisega [tavapäraste testidega], 16-18% patsientidest), paranenud histoloogiline pilt (38-52% patsientidest oli ≥ 2 punkti vähenemine vastavalt Knodelli histoloogilise aktiivsuse indeksile (HAI)) ja vähenenud fibroosi progresseerumine (3–17% patsientidest) ja tsirroos.
Pikaajaline ravi lamivudiiniga veel kaks aastat patsientidel, kellel ei õnnestunud esialgsetes üheaastastes kontrollitud uuringutes saavutada HBeAg serokonversiooni, näitas sidumisfibroosi edasist paranemist. HBV YMDD mutandiga patsientidel paranesid maksapõletiku parameetrid 41/82 (50%) patsiendil, 40/56 (71%) HBV YMDD mutandita patsientidel. Sildfibroos paranes 19/30 (63%) YMDD mutandita patsiendil ja 22/44 (50%) mutandiga patsiendil. Viis protsenti (3/56) YMDD mutandita patsiente ja 13% (11/82) YMDD mutandiga patsiente näitasid maksapõletiku parameetrite halvenemist võrreldes ravieelse olukorraga. Tsirroosi progresseerumine esines 4/68 (6%) YMDD mutandiga patsiendil, samas kui ühelgi ilma mutandita patsiendil ei olnud tsirroosi.
Aasia patsientidel (NUCB3018) laiendatud raviuuringus olid HBeAg serokonversiooni määr ja ALAT-i normaliseerumise määr 5-aastase raviperioodi lõpus 48% (28/58) ja 47% (15/32). HBeAg serokonversioon suurenes ALAT taseme tõusuga patsientidel: 77% (20/26) patsientidest, kelle ALAT oli> 2 ULN enne ravi, oli serokonversioon. 5 aasta lõpus oli kõigil patsientidel HBV DNA tase, mis oli kas tuvastamatu või alla ravieelse taseme.
Tabelis 2 on kokku võetud YMDD mutandi olemasolu põhjal levitatud uuringu täiendavad tulemused.
Tabel 2: 5 -aastane efektiivsus - tulemused, mis põhinevad YMDD mutandi olemasolul / puudumisel (Aasia uuring) NUCB3018
1. YMDD mutantidena märgiti need patsiendid, kelle HBV YMDD mutant oli ≥ 5% vähemalt ühel iga-aastasel testil 5-aastase perioodi jooksul. Mitte-YMDD mutantidena klassifitseeriti need patsiendid, kelle metsiku HBV viiruse protsent oli> 95% iga-aastased testid 5-aastase õppeperioodi jooksul.
2. normi ülempiirid
3. Abbott Genostics Solution Hybridization Test (LLOD)
4. Chiron Quantiplex test (LLOD 0,7 Meq / ml)
YMDD mutandi olemasolul põhinevad võrdlusandmed olid saadaval ka histoloogiliseks analüüsiks, kuid ainult kuni kolmeks aastaks. HBV YMDD mutandiga patsientidel paranes 18/39 (46%) nekro -põletikuline aktiivsus ja 9/39 (23 Mutandita patsientidel oli 20/27 (74%) nekro -põletikuline aktiivsus paranenud ja 2/27 (7%) halvenes.
Pärast HBeAg serokonversiooni on seroloogiline ravivastus ja kliiniline remissioon pärast lamivudiini kasutamise lõpetamist üldiselt kestvad. Siiski võib pärast serokonversiooni tekkida retsidiiv. Pikaajalises jälgimisuuringus esines patsientidel, kellel oli varasem serokonversioon ja lamivudiinravi katkestati, hilinenud viroloogiline retsidiiv. 39% patsientidest. Seetõttu tuleb pärast HBeAg serokonversiooni patsiente perioodiliselt jälgida, et hinnata seroloogiliste ja kliiniliste ravivastuste säilimist. Pikaajaline seroloogiline vastus säilib. HBV kliinilise kontrolli taastamiseks tuleks kaaluda korduvat ravi lamivudiini või alternatiivse viirusevastase ravimiga. .
Patsientidel, keda jälgiti kuni 16 nädalat pärast ravi lõpetamist ühe aasta jooksul, täheldati lamivudiinravi saanud patsientidel ALAT-i suurenemist pärast ravi sagedamini kui platseebot saanud patsientidel. Tabelis 3 on näidatud ravi järgsed ALAT taseme tõusud 52. ja 68. nädala vahel patsientidel, kes lõpetasid lamivudiini kasutamise 52. nädalal ja platseebot kogu ravikuuri vältel samades uuringutes. ravi ALAT tõus koos bilirubiini taseme tõusuga oli madal ja sarnane nii lamivudiini kui ka platseebot saanud patsientidel.
Tabel 3: ravi järgsed ALAT tõusud kahes täiskasvanute platseebokontrollitud uuringus
* Iga patsient võib olla esindatud ühes või mitmes kategoorias
† Võrreldav 3. astme toksilisusega vastavalt muudetud WHO kriteeriumidele
ULN = normi ülempiir
Kogemus CHB HBeAg negatiivsega patsientidel: Esialgsed andmed näitavad, et lamivudiini efektiivsus HBeAg -negatiivse CHB -ga patsientidel on sarnane HBeAg -positiivse CHB -ga patsientide omaga, kus 71% -l patsientidest oli HBV DNA supressioon alla testi tuvastamise piiri, 67% -l ALAT -i normaliseerus ja 38% -l paranes HAI pärast aastat ravist. Lamivudiini ärajätmisel ilmnes enamikul patsientidest (70%) viiruse replikatsiooni taastumine. Andmed on saadud pikaajalise ravi uuringust (NUCAB3017), mis hõlmas lamivudiiniga ravitud HBeAg negatiivseid patsiente.Pärast kaheaastast ravi selles uuringus esines ALAT normaliseerumine ja HBV DNA tuvastamatus vastavalt 30/69 (43%) ja 32/68 (47%) patsiendil, samas kui nekro -põletikulise skoori paranemist tõi esile 18/49 (37%) ) patsiendid. Patsientidel, kellel ei olnud HBV YMDD mutanti, täheldati 14/22 (64%) patsiendil nekro-põletikulise indeksi paranemist ja 1/22 (5%) patsiendi seisund oli halvenenud võrreldes ravieelse olukorraga. Mutandiga patsientidel esines 4/26 (15%) patsiendil nekro-põletikulise indeksi paranemine ja 8/26 (31%) patsiendi seisund halvenes võrreldes ravi-eelse olukorraga. Kumbki patsient ei jõudnud tsirroosini.
HBV YMDD mutandi hädaolukordade sagedus ja mõju ravivastusele: lamivudiini monoteraapia tulemusena valitakse HBV YMDD mutant umbes 24% patsientidest pärast üheaastast ravi, mis suureneb pärast 69 -aastast ravi 69% -ni. HBV YMDD mutandi areng on mõnedel patsientidel seotud ravivastuse vähenemisega mida tõestab HBV DNA taseme tõus ja ALAT tõus võrreldes varasema tasemega ravi ajal, hepatiidi nähtude ja sümptomite progresseerumine ja / või maksa nekroinflammatsiooni indeksite halvenemine. HBV YMDD mutandiga patsientide optimaalset ravijuhtimist ei ole veel kindlaks tehtud ( vt lõik 4.4).
Topeltpimedas uuringus, HBV YMDD mutantse CHB ja kompenseeritud maksahaigusega (NUC20904) patsientidel, kellel oli vähenenud viroloogiline ja biokeemiline ravivastus lamivudiinile (n = 95), lisati praegusel hetkel 10 mg adefoviirdipivoksiili 100 mg lamivudiini raviskeemi 52 nädala jooksul vähenes HBV DNA mediaan 4,6 log10 koopiat / ml, võrreldes ainult 0,3 log10 koopia / ml tõusuga ainult lamivudiiniga ravitud patsientidel. ALAT tasemed normaliseerusid 31% (14/45) kombineeritud ravi saanud patsientidest, võrreldes 6% (3/47) patsientidega, kes said ainult lamivudiini. Viiruse supressioon püsis (jätku-uuring NUC20917) kombineeritud raviga teisel raviaastal 104. nädalal, patsientidel jätkus viroloogilise ja biokeemilise ravivastuse paranemine.
Retrospektiivses uuringus HBV DNA tõusuga seotud tegurite kindlakstegemiseks raviti 159 HBeAg -positiivset Aasia patsienti lamivudiiniga ja jälgiti keskmiselt vähemalt 30 kuud. Patsientidel, kelle HBV DNA tase oli 6 kuud (24 nädalat) lamivudiinravi ajal suurem kui 200 koopiat / ml, oli YMDD mutandi tekke tõenäosus 60%, võrreldes 8% -ga madalama HBV DNA tasemega patsientidest. 24 nädalat lamivudiinravi YMDD mutandi tekke risk oli 63%, võrreldes 13% -ga, piirmääraga 1000 koopiat / ml (NUCB3009 ja NUCB3018).
Kogemus dekompenseeritud maksahaigusega patsientidel: platseebo-kontrollitud uuringuid ei tehtud dekompenseeritud maksahaigusega patsientidel, kuna neid peeti sobimatuteks. Kontrollimata uuringutes, kus lamivudiini manustati enne siirdamist ja selle ajal, ilmnes HBV DNA efektiivne supressioon ja ALAT normaliseerumine. Kui lamivudiinravi jätkati pärast siirdamist, vähenes siirdamise taasinfektsiooni määr HBV poolt, HBsAg kadu ja üheaastane elulemus 76-100%.
Nagu oodatud, oli samaaegse immunosupressiooni tõttu HBV YMDD mutantide esinemissagedus pärast 52 ravinädalat maksasiirdamise populatsioonis kõrgem (36% - 64%) kui immuunpuudulikkusega CHB patsientidel (14% - 32%).
Nelikümmend patsienti (HBeAg negatiivne või HBeAg positiivne), kellel oli kas dekompenseeritud maksahaigus või HBV retsidiiv pärast maksa siirdamist ja YMDD mutant, kaasati avatud uuringurühma NUC20904. 10 mg adefoviirdipivoksiili lisamine üks kord päevas praeguse lamivudiini raviskeemi kohaselt 100 mg 52 nädala jooksul näitas HBV DNA mediaanset langust 4,6 log10 koopiat / ml. Pärast üheaastast ravi paranes ka maksafunktsioon. Viiruse supressioon püsis (jätku-uuring NUC20917) kombineeritud ravi ajal teisel raviaastal 104. nädalal ning enamikul patsientidest paranesid maksafunktsiooni markerid ja nad said kliinilisest kasust kasu.
Kogemus kaugelearenenud fibroosi või tsirroosiga CHB -ga patsientidel: platseebokontrollitud uuringus, milles osales 651 kliiniliselt kompenseeritud kroonilise B-hepatiidi ja histoloogiliselt kinnitatud fibroosi või tsirroosiga patsienti, vähendas ravi lamivudiiniga (keskmine kestus 32 kuud) oluliselt haiguse üldist progresseerumist (34/436, 7,8% lamivudiini ja 38/ 215, 17,7% platseebo puhul, p = 0,001), mida näitab märkimisväärselt vähenenud nende patsientide osakaal, kellel oli suurenenud Child-Pugh väärtus (15/436, 3, 4% versus 19/215, 8,8%, p = 0,023) või kellel tekkis hepatotsellulaarne kartsinoom (17/436, 3,9% versus 16/215, 7,4%, p = 0,047). Üldine haiguse progresseerumise määr lamivudiinirühmas oli kõrgem HBV YMDD mutandiga patsientidel (23/209, 11%) kui neil, kellel HBV YMDD mutanti ei olnud (11/221, 5%). Haiguse progresseerumine YMDD mutantsetel isikutel lamivudiinirühmas oli siiski madalam kui haiguse progresseerumine platseeborühmas (vastavalt 23/209, 11% versus 38/214, 18%). Kinnitatud HBeAg serokonversiooni esines 47% (118/252) lamivudiiniga ravitud isikutest ja 93% (320/345) lamivudiini kasutanud isikutest muutus HBV DNA negatiivseks (VERSANT [versioon 1], bDNA test, LLOD
Kogemus lastel ja noorukitelLamivudiini manustati kompenseeritud CHB-ga lastele ja noorukitele platseebokontrollitud uuringus, milles osales 286 patsienti vanuses 2 ... 17 aastat. See populatsioon koosnes enamasti minimaalse B -hepatiidiga lastest. 2 ... 11 -aastastel lastel on kasutatud annust 3 mg / kg üks kord päevas (kuni 100 mg päevas) ja noorukitel vanuses 12 aastat või vanematel annustel 100 mg üks kord ööpäevas. Seda analüüsi tuleb täiendavalt kinnitada. Erinevus HBeAg serokonversiooni indeksites (HBeAg ja HBV DNA kadumine koos HBeAb tuvastamisega) platseeborühma ja lamivudiini rühmade vahel ei olnud selles populatsioonis statistiliselt oluline (indeksid ühe aasta pärast olid 13% (12/95) platseeborühmas) 22% (42/191) lamivudiinirühma puhul; p = 0,057). HBV YMDD mutandi esinemissagedus oli sarnane täiskasvanutel täheldatuga, vahemikus 19%, 52. nädalal kuni 45% pidevalt ravitud patsientidel 24 kuuks.
05.2 Farmakokineetilised omadused
Imendumine: Lamivudiin imendub seedetraktist hästi ja suukaudse lamivudiini biosaadavus täiskasvanutel on tavaliselt 80–85%. Pärast suukaudset manustamist on maksimaalse seerumi maksimaalse kontsentratsiooni (Cmax) saavutamise keskmine aeg (Tmax) ligikaudu 1 tund. Terapeutiliste annuste, st 100 mg / päevas korral on Cmax suurusjärgus 1,1-1,5 mcg / ml ja minimaalsed väärtused 0,015-0,020 mcg / ml.
Lamivudiini samaaegne manustamine koos toiduga põhjustab Tmax viivituse ja Cmax vähenemise (vähenenud kuni 47%). Kuna lamivudiini imendumise kiirus (AUC põhjal) ei muutu, võib lamivudiini manustada koos toiduga või ilma.
Jaotumine: Intravenoosse manustamise järgsed uuringud näitavad, et keskmine jaotusruumala on 1,3 l / kg. Lamivudiini farmakokineetika terapeutiliste annuste vahemikus on lineaarne ja plasma seondumine albumiiniga on väike.
Piiratud andmed näitavad, et lamivudiin siseneb kesknärvisüsteemi ja jõuab tserebrospinaalvedelikku. Keskmine suhe lamivudiini kontsentratsiooni vahel CSF-is ja seerumis 2-4 tundi pärast suukaudset manustamist on umbes 0,12.
Biotransformatsioon: Lamivudiin eritub muutumatul kujul peamiselt neerude kaudu. Piiratud metabolismi maksas (5-10%) ja vähenenud plasmavalkudega seondumise tõttu on teiste ainete metaboolsete koostoimete tõenäosus lamivudiiniga väike.
Eliminatsioon: Lamivudiini keskmine süsteemne kliirens on ligikaudu 0,3 l / h / kg. Keskmine täheldatud eliminatsiooni aeg on 5 kuni 7 tundi. Lamivudiin eritub peamiselt muutumatul kujul uriiniga glomerulaarfiltratsiooni ja aktiivse sekretsiooni (orgaaniline katioonide transpordisüsteem) kaudu. Neerukliirens moodustab 70% lamivudiini eliminatsioonist.
Patsientide erikategooriad: Neerupuudulikkusega patsientidega läbi viidud uuringud näitavad, et neerupuudulikkus mõjutab lamivudiini eliminatsiooni. Patsientidel, kelle kreatiniini kliirens on alla 50 ml / min, tuleb annust vähendada (vt lõik 4.2).
Maksafunktsiooni kahjustus ei mõjuta lamivudiini farmakokineetikat. Piiratud andmed maksasiirdamise patsientide kohta näitavad, et maksa dekompensatsioon ei mõjuta oluliselt lamivudiini farmakokineetikat, kui sellega ei kaasne neerufunktsiooni häireid.
Lamivudiini farmakokineetilise profiili põhjal on mõeldav, et eakatel patsientidel ei avalda normaalne vananemine koos neerufunktsiooni langusega olulist kliinilist mõju lamivudiini ekspositsioonile, kui välistada patsiendid, kelle kreatiniini kliirens on alla 50 ml / min (vt lõik 4.2).
05.3 Prekliinilised ohutusandmed
Loomkatsetes ei seostatud lamivudiini suurte annuste manustamisega olulist toksilisust elunditele. Suuremate annuste kasutamisel täheldati väikest mõju maksa- ja neerufunktsiooni näitajatele ning aeg -ajalt maksa kaalu langust.
Tõenäoliselt on kliiniliselt kõige olulisemaks toimeks erütrotsüütide ja neutrofiilide arvu vähenemine, millest kliinilistes uuringutes teatati harva.
Lamivudiin ei olnud bakteritestides mutageenne, kuid sarnaselt paljudele nukleosiidi analoogidele näitas tsütogeneetilises testis aktiivsust in vitro ja hiire lümfoomi testis. Lamivudiin ei ole genotoksiline in vivo annustes, mis indutseerivad plasmakontsentratsiooni ligikaudu 60-70 korda kõrgemal kui kliinilises keskkonnas oodatud plasmatasemed. Nagu mutageenne toime in vitro lamivudiini sisaldust testidega ei kinnitatud in vivo, sellest järeldub, et lamivudiin ei põhjusta eeldatavasti ravi saavatel patsientidel genotoksilist riski.
Reproduktsiooniuuringud loomadel ei ole näidanud teratogeensust ega mõju isaste või emaste viljakusele.Kui tiinetele küülikutele manustatakse lamivudiin inimestel saavutataval tasemel, põhjustab see embrüo varase surma. Seda ei esine rottidel isegi väga suure süsteemse ekspositsiooni korral.
Lamivudiini pikaajaliste kartsinogeensusuuringute tulemused rottidel ja hiirtel ei näidanud kantserogeenset toimet.
06.0 FARMATSEUTILINE TEAVE
06.1 Abiained
Sahharoos (20% mass / maht)
Metüülparahüdroksübensoaat (E218)
Propüülparahüdroksübensoaat (E216)
Sidrunhape (veevaba)
Propüleenglükool
Naatriumtsitraat
Kunstlik maasika maitse
Kunstlik banaani maitse
Puhastatud vesi
06.2 Sobimatus
Ei ole asjakohane.
06.3 Kehtivusaeg
2 aastat.
Pärast esmast avamist: 1 kuu
06.4 Säilitamise eritingimused
Hoida temperatuuril mitte üle 25 ° C
06.5 Vahetu pakendi iseloomustus ja pakendi sisu
Pakend, mis sisaldab 240 ml lamivudiini suukaudset lahust läbipaistmatus valges suure tihedusega polüetüleenist (HDPE) pudelis koos lastekindla polüpropüleenist korgiga. Pakend sisaldab ka polüetüleensüstla adapterit ja 10 ml suukaudset doseerimissüstalt, mis koosneb silindrilisest polüpropüleenist korpusest (ml astmetega) ja polüetüleenkolbist.
Suukaudne doseerimissüstal on ette nähtud koguse suukaudse lahuse täpseks annustamiseks. Kasutusjuhend on pakendile lisatud.
06.6 Kasutamis- ja käsitsemisjuhised
Kasutamata ravim tuleb hävitada vastavalt kohalikele eeskirjadele.
07.0 MÜÜGILOA HOIDJA
Glaxo Group Ltd
980 Great West Road
Brentford
Middlesex
TW8 9GS
Ühendkuningriik
08.0 MÜÜGILOA NUMBER
EL/1/99/114/003
034506030
09.0 MÜÜGILOA VÕI UUENDAMISE KUUPÄEV
Müügiloa esmase väljastamise kuupäev: 29. juuli 1999
Viimase uuendamise kuupäev: 27. august 2009
10.0 TEKSTI LÄBIVAATAMISE KUUPÄEV
Jaanuar 2014











-cos-esami-e-terapia.jpg)