Aperti sündroom on täheldatav igal 68 000-88 000 vastsündinul, kuna see on tingitud FGFR2 geeni spetsiifilisest mutatsioonist, mille ülesandeks on reguleerida koljuõmbluste sulandumist ning sõrmede ja varvaste arengut.
Aperti sündroomi diagnoosimisel on oluline füüsiline läbivaatus, anamnees, kolju ja sõrmede ja varvaste radioloogiline hindamine ning lõpuks geneetiline test.
Praegu saavad Aperti sündroomi all kannatavad inimesed loota ainult sümptomaatilisele ravile, see tähendab, et nad leevendavad sümptomeid ja väldivad kõige tõsisemaid tüsistusi.
Lühike ülevaade koljuõmblustest ja nende sulandumisest
Kraniaalõmblused on kiulised liigesed, mis ühendavad kraniaalvõlvi luid (st eesmise, ajalise, parietaalse ja kuklaluu).
Normaalsetes tingimustes toimub koljuõmbluste sulandumisprotsess sünnijärgsel perioodil, alustades mõnede liigeste elementide puhul 1–2-aastasest ja lõpetades 20-aastaselt. See pikk ja järkjärguline sulandumisprotsess võimaldab ajul piisavalt kasvada ja areneda.
Aperti sündroom on aga tänu oma tuntusele mitte ainult seotusega kraniostenoosiga, vaid ka asjaoluga, et see on seotud teatud sündaktüüli astmega, see tähendab kaasasündinud anomaaliaga, mida iseloomustab ühe või mitme sõrme või sõrme liitmine. jalad.
Võimalus samaaegselt põhjustada kraniostenoosi ja sündaktüüli muudab Aperti sündroomi akrotsefalosündaktüüli näiteks; meditsiinis on "akrotsefaalosündaktiline" geneetiline seisund, mis ühendab kolju spetsiifilised väärarengud ("acrocephalus" tähendab "peast otsani") ühe või mitme sõrme või varba sulandumisega.
Millised on varajase koljuõmbluse liitmise tagajärjed?
Kui koljuõmblused sulanduvad sünnieelse, perinataalse (*) või väga varajase lapsepõlve ajal, nagu Aperti sündroomi ja muude sellega seotud haiguste puhul, siis ajuorganid, nagu aju, väikeaju ja ajutüvi ning silmade läbimisel muutused nii kasvus kui ka vormis.
* NB! "Perinataalne elu" tähistab ajavahemikku 27. rasedusnädala ja esimese 28 päeva jooksul pärast sünnitust.
Epidemioloogia: kui levinud on Aperti sündroom?
Statistika kohaselt sünnib iga 65 000-88 000 inimest Aperti sündroomiga.
Kas teadsite, et ...
Geneetilised haigused, mis nagu Aperti sündroom, põhjustavad kraniosünostoosi, on umbes 150.
Nende hulgas tõusevad lisaks Aperti sündroomile esile Crouzoni sündroom, Pfeifferi sündroom ja Saethre-Chotzeni sündroom.
Uudishimu
Omandatud mutatsioon, mis põhjustab Aperti sündroomi, on näide "mutatsioonist" de novo", st" uuest mutatsioonist, millel puudub pärilik olemus ".
Mis põhjustab Aperti sündroomiga seotud geenimutatsiooni?
Eeldus: inimese kromosoomidel esinevad geenid on DNA järjestused, mille ülesanne on toota eluks olulistes bioloogilistes protsessides, sealhulgas rakkude kasvus ja replikatsioonis, põhilisi valke.
Kui Aperti sündroomiga seotud FGFR2 geen on mutatsioonivaba (seega terves inimeses), toodab see õiges koguses retseptorvalku, mida nimetatakse fibroblastide kasvufaktori retseptoriks 2, mis on hädavajalik kraniaalse sulandumise ajastamiseks. õmblusniidid ning sõrmede ja varvaste eraldumise jälgimine (teisisõnu, see annab märku, millal on õige aeg koljuõmbluste sulandumiseks ning reguleerib sõrmede ja varvaste teket).
Teisest küljest, kui FGFR2 geen läbib Aperti sündroomi juuresolekul täheldatud mutatsiooni, on see hüperaktiivne ja toodab ülalnimetatud retseptorvalku sellistes massiivsetes kogustes, et koljuõmbluste sulandumisega seotud ajastus muutub (see on kiiremini) ning sõrmede ja varvaste eraldamise protsessid ei toimu õigesti.
Kes on kõige rohkem ohus?
Mis puudutab omandatud Aperti sündroomi juhtumeid, siis faktorid, mis indutseerivad FGFR2 geeni mutatsiooni pärast eostamist, pole praegu täiesti selged.
Selle aspekti uuringud jätkuvad.
Aperti sündroom on autosoomne domineeriv haigus
Aru saama...
Iga inimese geeni esineb kahes eksemplaris, mida nimetatakse alleelideks, millest üks on ema ja teine isapoolne.
Aperti sündroomil on kõik autosomaalse domineeriva haiguse tunnused.
Geneetiline haigus on autosomaalselt domineeriv, kui seda põhjustava geeni ühe eksemplari mutatsioon on piisav avaldumiseks.
- Lame või nõgus nägu (näo kesksete luude puuduliku kasvu tõttu)
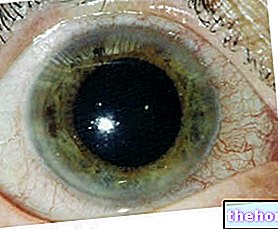
- Tursed, punnis ja pärani avatud silmad madalad silmakoopad ja ebanormaalselt asetsevad silmad (silmakoopade hüpertelorism);
- Nokaga nina;
- Arenenud lõualuu, kombineerituna prognatismiga;
- Ülerahvastatud hambad (vähearenenud lõualuu tõttu)
- Kõrvad tavalisest madalamad.
Sündaktiilselt
Aperti sündroomi kandjatel täheldatakse sündaktüüli kätes, peaaegu alati ja jalgades, harvem kui kätes.

Aperti sündroomiga inimese käes on sündaktüüli tüüpilised omadused 4:
- Radiaalsete kõrvalekalletega lühikese pöidla olemasolu (st orienteeritud ebanormaalselt raadiusele, üks käsivarre kahest luust);
- Kompleksne sündroomia nimetissõrme, keskmise sõrme ja sõrmuse sõrme vahel. Kompleksse sündaktüüli all peavad arstid silmas sõrmede ebanormaalset liitumist, mis mõjutab mitte ainult pehmeid kudesid (nahka), vaid ka luukoe (falange);
- Sümfanglus. See on meditsiiniline termin, mis näitab sõrmede interfalangeaalsete liigeste anomaalset sulandumist (interfalangeaalliigesed on falanksi ja falanksi vahel asuvad liigeseelemendid);
- Lihtne sündaktia neljanda ja viienda varba vahel (st sõrmuse ja väikeste sõrmede vahel). Lihtsa sündaktüüli puhul viitavad eksperdid sõrmede ebanormaalsele liitumisele, mis mõjutab ainult pehmeid kudesid (nahka).
SÜNDROOMI TÕSISUS AVATUD SÜNDROOMIS: 3 TÜÜPI
Pöidla väärarengu raskusastme (esimene neljast tunnusest) põhjal eristavad Aperti sündroomi eksperdid kolme järjest suureneva raskusastmega sündaktüüli:
- I tüüp (kõige vähem tõsine) langeb kokku "pöialt mõjutava minimaalse anomaaliaga, mis jääb indeksist täiesti sõltumatuks".
Muud anomaaliad: nimetissõrm, keskmine ja sõrmusesõrm on kokku sulatatud kompleksse sündaktüüli kaudu ja esinevad sümpalangismid, mis mõjutavad distaalseid interfalangeaalseid liigeseid; c "on lihtne ja mittetäielik sündaktiaalsus sõrmuse ja väikeste sõrmede vahel (mittetäielik sündaktilus tähendab, et kahe sõrme sulandumine on osaline).
Muu info: on kõige levinum tüüp. - II tüüpi (keskmise raskusastmega) iseloomustab pöidla radiaalsem kõrvalekalle võrreldes eelmise juhtumiga ning pöidla ja nimetissõrme vahelise sündaktilise põhimõtte järgimine (c "on mittetäielik sündikaalia pöidla ja nimetissõrme vahel). .
Muud anomaaliad: nimetissõrm, keskmine ja sõrmusesõrm on peategelased kompleksse sündaktiliselt kombineeritud distaalse sümplangismiga; sõrmusesõrme ja väikese sõrme vahel c "on lihtne ja mittetäielik sündaktiil.
Muu info: see on teine levinum tüüp. - III tüüpi (kõige raskemat) iseloomustab pöidla olemasolu, mis on indeksiga täielikult ühendatud mitte ainult pehmete kudede, vaid ka luukoe tasemel.
Muud anomaaliad: kõik sõrmed on kokku sulanud, nii et neid on peaaegu võimatu ära tunda; c "on" üksik nael; kui esimese nelja sõrme vahel on sündaktiil keeruline, siis sõrmusesõrme ja väikese sõrme vahel on see (nagu ka teiste tüüpide puhul) lihtne ja mittetäielik.
Muu info: see on kõige haruldasem tüüp.
Aperti sündroomi muud võimalikud sümptomid ja tunnused
Mõnel juhul on Aperti sündroom lisaks kraniosünostoosile ja sündaktüülile seotud ka järgmiste teguritega: polüdaktüülia (st käe või jala liigse sõrme olemasolu), kuulmislangus, kõrvade ja siinuste kordumine, hüperhidroos, rasune nahk, raske akne, karvade puudumine kulmudel, kaelalülide liitmine, obstruktiivne uneapnoe sündroom ja / või suulaelõhe.
Tüsistused
Aperti sündroomi tüsistused on ennekõike tõsised tagajärjed, mis kraniosünostoosil võivad olla aju ja intellektuaalsete võimete arengule ning sündaktüüli alluvate käte funktsionaalsetele võimalustele.
Millal on võimalik avastada Aperti sündroomi?
Tavaliselt ilmnevad Aperti sündroomist tingitud kolju- ja digitaalsed kõrvalekalded sündides, seega on diagnoosimine ja ravi planeerimine kohene.
pea (pea röntgenikiirgus, pea kompuutertomograafia ja / või pea MRI) ning käed ja võib-olla jalad; lõpuks lõpeb see geneetilise testiga.
Füüsiline läbivaatus ja haiguslugu
Füüsiline läbivaatus ja anamnees seisnevad peamiselt patsiendi sümptomite täpses uurimises.
Aperti sündroomi kontekstis leiab arst nendel diagnoosimisprotsessi hetkedel kraniosünostoosi ja sündaktüüli ning nende täpsed omadused.
Pea, sõrmede ja varvaste radioloogilised uuringud
Aperti sündroomi kontekstis:
- Arst kasutab pea radioloogilisi uuringuid, et kinnitada koronaalsete õmbluste varajase sulandumise olemasolu (koronaalne kraniosünostoos või brahütsefaalia); pealegi võimaldavad need tal hinnata praeguste kranio-entsefaalsete anomaaliate tõsidust.
- Teisest küljest on sõrmede ja varvaste radioloogilised uuringud hädavajalikud mitte niivõrd sündikaalia kinnitamiseks (selleks piisab visuaalsest vaatlusest), vaid pigem selleks, et üksikasjalikult teada saada interdigitaalsete fusioonide omadusi (sündikaali tüüp, tase termotuumasünteesi jne).
Geneetiline test
See on DNA analüüs, mille eesmärk on tuvastada kriitiliste geenide mutatsioone.
Aperti sündroomi kontekstis kujutab see kinnitavat diagnostilist testi, kuna toob päevavalgele kõnealusele geneetilisele haigusele iseloomuliku FGFR2 mutatsiooni.
BRACHYCEPHALIA KIRURGILINE HOOLDUS
Aperti sündroomi kandja jaoks hõlmab brahütsefaalia kirurgiline ravi järgmist:
- Esimene sekkumine noores eas (eluaasta jooksul), mille eesmärk on eraldada koronaalsed õmblusniidid oodatust varem. Kui see sekkumine õnnestub, on ajul õige kasvuruum ja väheneb intellektuaalsete probleemide oht.
- Teine sekkumine vanuses 4 kuni 12 aastat, mille eesmärk on anda näole normaalne välimus, mis (nagu lugeja mäletab) on tasane, kui mitte nõgus.
Kõnealune operatsioon hõlmab teatud näo luude sisselõikamist ja nende ümberpaigutamist vastavalt korrale, mis vähemalt osaliselt peegeldab normaalsust. - Kolmas võimalik sekkumine lapsepõlves, eesmärgiga kõrvaldada või vähemalt vähendada silma hüpertelorismi.
SÜNDAKSIA KIRURGILINE HOOLDUS
Sündaktiilne kirurgiline ravi varieerub vastavalt interdigitaalse sulandumise omadustele (seega sõltub see tüübist).

See tähendab, et Aperti sündroomiga indiviidi puhul kehtiv sekkumine ei pruugi olla sama kehtiv teise sama geneetilise haigusega isiku puhul (see kehtib ainult siis, kui sündaktüüli tüüp on sama).
Olles selgitanud seda põhiaspekti, on iga olemasoleva kirurgilise lähenemisviisi eesmärk sama ja koosneb sulatatud sõrmede vabastamisest, et tagada kätele teatud funktsionaalsus.
Üldiselt hõlmab sündaktüütiline ravi kahte etappi:
- 1 samm: "vabastage" esimene interdigitaalne ruum (pöidla ja nimetissõrme vahe) ja neljas interdigitaalne ruum (tühiku ja väikese sõrme vahe);
- 2. samm: "vabastage" teine ja kolmas interdigitaalne ruum (tühik nimetissõrme ja keskmise sõrme vahel ning tühi keskmise ja sõrmuse sõrme vahel).