Üldisus
Osteogenesis imperfecta on kaasasündinud geneetiline haigus, mis ei ole seotud sooga, põhjustades teatud luude haprust ja luumurdude kalduvust.
Osteogeneesi imperfecta sümptomeid on palju; üldiselt koosnevad need: luude nõrgenemisest, suurest kalduvusest luumurdudele, sinise, halli või lilla silmapõletiku olemasolust, luude deformatsioonidest või muudest skeleti muutustest, kolmnurksest näost, hammaste haprusest jne. .
Üldiselt on osteogenesis imperfecta õigeks diagnoosimiseks hädavajalik järgmine: füüsiline läbivaatus, haiguslugu, meditsiinilised pilditestid, I tüüpi kollageeni hindamise test ja geneetiline test.
Kahjuks on praegu osteogenesis imperfecta patsientidele kättesaadavad ainsad ravimeetodid sümptomaatilised. Kõnealune haigus on tegelikult ravimatu.
Mis on osteogenesis imperfecta?
Osteogenesis imperfecta on geneetiline haigus, mis muudab haige inimese luud nõrgemaks ja altid luumurdudele.
Tegelikkuses viitavad arstid terminiga osteogenesis imperfecta heterogeensele geneetiliste haiguste rühmale, mida iseloomustab teatud määral luude haprus. Seetõttu on osteogenesis imperfecta mitmeid vorme (või tüüpe), millest mõned on palju raskemad kui teised.
See on kaasasündinud haigus
Inimestel, keda see mõjutab, on osteogenesis imperfecta haigus, mis esineb sünnist saati, seega võib seda igati määratleda kaasasündinud haigusena.
KAS SEKS ON SEOTUD?
Osteogenesis imperfecta ei ole sooga seotud geneetiline haigus, näiteks hemofiilia või Klinefelteri sündroom.
EPIDEMILOOGIA
Mõnede statistiliste uuringute kohaselt oleks osteogenesis imperfecta esinemissagedus võrdne ühe juhtumiga iga 15 000–20 000 sünnituse kohta. See tähendab, et igal 15 000–20 000 vastsündinul on üks, keda mõjutab osteogenesis imperfecta.
Teised statistilised uuringud on samuti näidanud, et osteogenesis imperfecta mõjutab mehi ja naisi võrdselt ning see ei eelista konkreetset elanikkonda ega rahvusrühma.
Eluiga on äärmiselt muutuv parameeter, mis sõltub osteogeneesi imperfecta vormist.
Põhjused
Osteogenesis imperfecta tuleneb peaaegu alati I tüüpi kollageeni tootmise kvalitatiivsest ja kvantitatiivsest muutumisest.
I tüüpi kollageen on hädavajalik luude tugevdamiseks ja kõhre, kõõluste, naha, silma sklera jne moodustavate tervislike sidekoe säilitamiseks.
Seetõttu mõjutab I tüüpi kollageeni tootmise muutus luude tugevust ja inimkehas esinevate sidekoe head tervist.
Mis muudab kollageeni tootmist?
Geneetiline haigus on seisund, mis tekib ühe või mitme raku DNA moodustava geeni mutatsiooni tõttu.
Osteogenesis imperfecta puhul on viimase põhjused peaaegu alati leitavad ühe või mõlema geeni COL1A1 (asub kromosoomis 17) ja COL1A2 (asub kromosoomis 7) mutatsioonis.
Normaalsetes tingimustes reguleerivad COL1A1 ja COL1A2 I tüüpi kollageeni normaalset tootmist; kui nende laengus on mutatsioone, ei suuda nad oma regulatiivset funktsiooni täita.
Tähtis: millised muud geenid, kui need on muteerunud, põhjustavad osteogenesis imperfecta?
Lisaks COL1A1 ja COL1A2 mutatsioonidele on mutatsioonid IFITM5, SERPINF1, CRTAP ja LEPRE1 geenides potentsiaalsed osteogenesis imperfecta põhjused.
Eespool nimetatud geenid hõlmavad funktsioone, mis erinevad COL1A1 ja COL1A2 funktsioonidest - seega ei kontrolli nad I tüüpi kollageeni tootmist -, kuid neil on siiski "mõju inimese luustiku luude tugevusele ja vastupidavusele.
MIS GENEETILINE HAIGUS SEE ON?
Osteogenesis imperfecta on autosoomne geneetiline haigus.
Termin autosomaalne, mis on seotud geneetilise haigusega, näitab, et kõnealune seisund on tingitud geneetilistest mutatsioonidest, mis põhinevad autosomaalsetes ja mitte-soolistes kromosoomides.
Lugejatele tuletatakse meelde, et inimesel on 23 paari kromosoome sisaldav kromosoomikomplekt, milles 22 paari on autosomaalseid ja ainult üks paar on seksuaalse tüübi. Seksuaaltüüpi kromosoomipaar mõjutab sugu individuaalne.
Osteogenesis imperfecta pärast mutatsioone COL1A1, COL1A2 ja IFITM5 omab kõiki autosomaalse domineeriva haiguse tunnuseid. Kui see on tingitud mutatsioonidest geenides SERPINF1, CRTAP ja LEPRE1, on sellel autosomaalse retsessiivse haiguse tunnused.
TÜÜBID
Praegu usuvad arstid, et osteogenesis imperfecta on 8 tüüpi (või vormi). Erinevate tüüpide eristamiseks otsustasid nad kasutada Rooma numeratsiooni, täpsemalt kaheksa esimest Rooma numbrit.
Allolev tabel näitab 8 osteogeneesi imperfecta vormi, neid põhjustavaid mutatsioone ja muid omadusi.
Kutt
Muteerunud geen
Geneetilise haiguse tüüp
THE
COL1A1
Autosomaalne dominant
II
COL1A1 ja COL1A2
Autosomaalne dominant
III
COL1A1 ja COL1A2
Autosomaalne dominant
IV
COL1A1 ja COL1A2
Autosomaalne dominant
V.
IFITM5
Autosomaalne dominant
SINA
SERPINF1
Autosomaalne retsessiivne
VII
CRTAP
Autosomaalne retsessiivne
VIII
HARE 1
Autosomaalne retsessiivne
* NB! Ilmselgelt on mutatsioonid COL1A1 ja COL1A2, mis põhjustavad nelja esimest osteogeneesi imperfecta vormi, geneetilised muutused, millel on veidi erinevad omadused. Vastasel juhul poleks mõtet üht teist eristada.
Sümptomid, sümptomid ja tüsistused
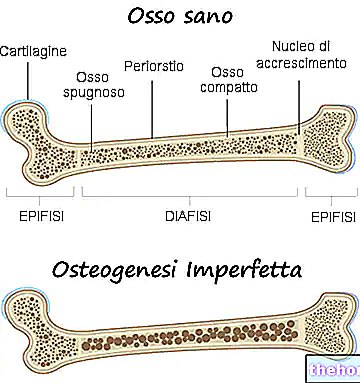
Igasugused osteogenesis imperfecta tüübid vastutavad luude nõrgenemise eest, nii et haigusest mõjutatud isikul on luumurdude suhtes eelsoodumus. Luude nõrgenemise aste varieerub sõltuvalt kujust; mõnel neist on see nõrgenemine suurem kui teistel.
Seda öeldes tuleb märkida, et igal osteogenesis imperfecta vormil on oma sümptomaatiline pilt, mis mõne jaoks võib meenutada teiste vormide sümptomaatilist pilti.
VÕIMALIKUD SÜMPTOMID JA MÄRGID
Osteogeneesi imperfecta võimalikud sümptomid ja tunnused on järgmised:
- Luu väärarengute olemasolu;
- Lühikese ja väikese keha olemasolu (mõeldud pagasiruumiks);
- Liigeste probleemid (nt lahtised liigesed);
- Lihaste nõrkus;
- Sinine, lilla või hall silma sklera;
- Kolmnurkne nägu;
- Tünn rind;
- Lülisamba morfoloogilised kõrvalekalded;
- Hammaste haprus;
- Kuulmise vähenemine või täielik kaotus;
- Hingamisprobleemid
- 1. tüüpi kollageeni puudumise või puudumisega seotud probleemid.
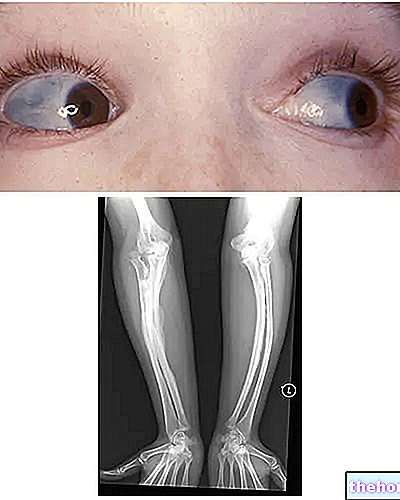
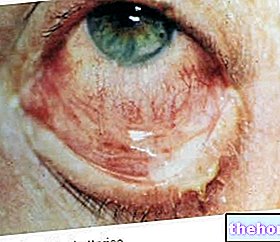
Osteogenesis Imperfecta: pange tähele sklera sinist värvi ja haigust iseloomustavaid luude deformatsioone. Alates wikipedia.org
MILLISED ON IMELISE OSTEOGENEESI TÕSISIMAD VORMID?
Arstid liigitavad erinevate osteogeneesi imperfecta tüüpide sümptomatoloogilise raskusastme skaalal 3 kraadi, mis on: kerge, keskmine ja raske.
Ainult üks vorm kuulub "kerge astme" kategooriasse: "I tüüpi osteogenesis imperfecta"; 4 osteogenesis imperfecta vormi kuuluvad "mõõduka astme" kategooriasse: IV, V ja VI; lõpuks kuuluvad "raske astme" kategooriasse 3 vormid: II, III, VII ja VIII.
I TÜÜP: OMADUSED
Kõige tavalisem ja kõige raskem vorm I tüüpi osteogenesis imperfecta on järgmised omadused:
- See põhjustab luumurde, eriti enne puberteeti;
- Sellel pole "peaaegu mingit mõju kõrgusele, seega on patsiendid tavaliselt normaalse pikkusega";
- Põhjustab liigeseprobleeme ja lihaste nõrkust
- See vastutab sinise, lilla või halli sklera eest;
- See on kolmnurkse näo ja selgroo anomaaliate põhjus;
- See ei põhjusta peaaegu kunagi luude deformatsioone. Kui see neid provotseerib, on need minimaalsed;
- See võib põhjustada hammaste haprust ja / või kuulmislangust (viimane esineb tavaliselt täiskasvanueas);
- Seda seostatakse I tüüpi kollageeni olemasoluga, mille kvaliteet on normaalne, kuid koguses ebanormaalne (see on tavalisest kehvem).
II TÜÜP: OMADUSED
II tüüpi osteogenesis imperfecta iseloomustab:
- Surma põhjus sünnil või varsti pärast seda. Hingamisprobleemid põhjustavad peaaegu alati surma;
- Märkimisväärne luude haprus ja tõsised luude deformatsioonid;
- Lühike kasv ja vähearenenud kopsud
- Sinine, lilla või halli värvi sklera;
- I tüüpi kollageeni kvantitatiivsete ja kvalitatiivsete kõrvalekallete olemasolu.
III TÜÜP: OMADUSED
III tüüpi osteogenesis imperfectal on järgmised omadused:
- Kuigi see on väga tõsine, ei põhjusta see sageli vastsündinute perioodil surma;
- Seda seostatakse "luude kõrge haprusega;
- See vastutab lühikese kasvu, liigeseprobleemide, lihaste nõrkuse (eriti jalgades ja kätes), tünnirinna, kolmnurkse näo ja selgroo ebanormaalse kõveruse eest;
- Selle põhjuseks on sinine, lilla või hall sklera;
- See võib põhjustada hingamisprobleeme, hammaste haprust ja kuulmislangust;
- See on sageli vastutav luude deformatsioonide eest;
- Seda seostatakse I tüüpi kollageeni kvalitatiivsete ja kvantitatiivsete kõrvalekalletega.
IV TÜÜP: OMADUSED
IV tüüpi osteogeneesi iseloomustavad:
- II ja III vormi ning vormi I luude hapruse aste;
- Keskmisest lühem kasv;
- Sinine, lilla või halli värvi sklera;
- Kerge / mõõduka keha luude deformatsioonid, selgroo ja tünni rindkere väikesed kõrvalekalded;
- Kolmnurkne nägu;
- Võimalik hammaste haprus ja kuulmislangus;
- I tüüpi kollageeni kõrvalekallete olemasolu.
V TÜÜP: OMADUSED
V tüüpi osteogenesis imperfecta sarnaneb mõnes mõttes IV tüüpi osteogenesis imperfecta'ga. Sellel on siiski mõned eripärad, näiteks:
- Normaalse värvusega sklera;
- Hammaste hapruse puudumine;
- Luude luumurdude paranemisprotsessi ajal ebanormaalsete kondiliste kalluste moodustumine;
- Raadiuse ja küünarluu vahel paikneva vaheseina membraani lubjastumine. See kahjustab küünarvarre liikuvust.
VI TÜÜP: OMADUSED
Ka VI tüüpi osteogenesis imperfecta sarnaneb vormiga IV. Selle eristamiseks viimasest on mõned eripärad, sealhulgas leeliselise fosfataasi kõrge sisaldus veres ja mõnede luude puhul lammaste (kondised) olemasolu, mis sarnanevad kala selgrooga.
VII TÜÜP: OMADUSED
Sümptomaatiliselt võib VII tüüpi osteogenesis imperfecta mõnel juhul sarnaneda IV tüübiga ja muudel juhtudel II tüübiga.
Selle tõsise patoloogilise vormi iseärasused on järgmised:
- Lühike kasv;
- Äärmiselt lühikese õlavarreluu (käeluu) ja reieluu (reieluu) olemasolu;
- Puusa deformatsiooni sagedane esinemine, mida tuntakse kui coxa vara.
VIII TÜÜP: OMADUSED
VIII tüüpi osteogenesis imperfecta meenutab väga II ja III vormi.
Omapäraste omaduste hulgas eristuvad järgmised: tõsine kasvupuudus, tugev skeleti hüpomineralisatsioon ja prolüül-3-hüdroksülaasi ensüümi puudumine (või napp esinemine).
Diagnoos
Üldiselt algab diagnoosimisprotsess, millega alustatakse kahtlustatava osteogenesis imperfecta vormiga patsiente, hoolika füüsilise läbivaatuse ja hoolika haiguslooga; seejärel jätkub see "patsiendi perekonna ajaloo analüüsiga ja diagnostiliste pilditestide seeriaga (röntgen, CT skaneerimine jne); lõpuks lõpeb see I tüüpi kollageeni kvantitatiivse ja kvalitatiivse hindamisega ning geneetiline test.
Tänapäeval on võimalus diagnoosida osteogenesis imperfecta isegi sünnieelses faasis, lastes rase ultraheli.
EESMÄRGILISE UURINGU JA AJALUGU TÄHTIS
Arst, osteogenesis imperfecta ekspert, suudab väga sageli diagnoosida ülalnimetatud haigust isegi ainult füüsilise läbivaatuse ja anamneesi abil. See tähendab, et need diagnostilised testid ei ole tähtsusetu.
I TÜÜBI KOLLAGENITOOTMISE HINDAMINE
Reeglina on I tüüpi kollageeni kvalitatiivne ja kvantitatiivne hindamine väga usaldusväärne test, sest nagu öeldud, on enamikul osteogenesis imperfecta juhtudest iseloomulikud mutatsioonid geenides, mis kontrollivad 1. tüüpi kollageeni tootmist.
Inimese rakutasandil esineva I tüüpi kollageeni koguse ja kvaliteedi hindamiseks võivad arstid tugineda nahabiopsiale või konkreetsele vereanalüüsile.
Mõlemad hindavad testid on üsna keerulised ja patsient (või tema vanemad) võib tulemuste teadasaamiseks oodata mitu nädalat.
GENEETILINE TEST
Läbi geneetilise testi, mis uurib uuritava inimese kogu DNA -d, saavad arstid lõplikult otsustada olemasoleva geneetilise mutatsiooni omaduste kohta.
Üldiselt on geneetilise testi tegemine kogu raku DNA jaoks ette nähtud, kui I tüüpi kollageeni omaduste hindamine ei ole andnud soovitud tulemusi või kui see ei ole mutatsioon COL1A1 või COL1A2, mis põhjustab "osteogenesis imperfecta".
PRENATAALDIAGNOOS
Sünnieelne ultraheli on väga kasulik II ja III tüüpi osteogenesis imperfecta tuvastamiseks.
Teraapia
Praegu pole spetsiifilist ravi osteogenesis imperfecta jaoks. Teisisõnu, osteogenesis imperfecta -ga inimesed on määratud elama ülalmainitud seisundiga kuni surmani, mis on sageli tingitud haiguse enda tagajärgedest.
Spetsiifilise ravi puudumine ei välista teiste ravivormide olemasolu. Tegelikult on osteogenesis imperfecta -ga patsiendi ravivõimaluste hulgas ka erinevaid sümptomaatilisi ravimeetodeid; sümptomaatilise ravi all peame silmas ravi, mis on võimeline sümptomeid leevendama, haiguse kulgu aeglustama ja kõige tõsisemaid tagajärgi ära hoidma (või vähemalt edasi lükkama).
VÕIMALIKUD SÜMPTOMAATSED RAVIMID
Osteogenesis imperfecta võimalike sümptomaatiliste ravimeetodite loetelus eristuvad järgmised:
- Küünte kirurgiline sisestamine kõige pikemate luude sisse (NB: kõige tõenäolisem murdumiskoht), mis tagab suurema vastupidavuse luumurdudele ja deformatsioonidele. Seda operatsiooni nimetatakse roodimine intramedullaarne;
- Luumurdude ja / või luude deformatsioonide konservatiivne või kirurgiline ravi;
- Hambaravi hammaste tervise kaitsmiseks;
- Valu leevendavad ravimeetodid väga valulike hulgimurdude korral;
- Füsioteraapia, lihaste pikendamiseks ja tugevdamiseks.Elastne ja tooniline lihaste aparaat võimaldab vältida kukkumisi, mis võivad põhjustada erinevaid luumurde;
- Liikumisabivahendite kasutamine, sealhulgas ratastoolid, traksid, kargud jne.
LIIKUMISE KASU
Osteogenesis imperfecta'ga inimestele soovitavad arstid pidevalt harjutada füüsilisi harjutusi ja üldiselt liikuda, kuna mõlemad tegevused aitavad kaasa luu- ja lihasüsteemide tugevdamisele.
Soovitatavate spordialade hulka kuuluvad: ujumine, kuna see on "vähese mõjuga füüsiline aktiivsus luustikule", ja kõndimine.
KASU TERVISEST ELUSTIILIST
Tervisliku eluviisi juhtimine, suitsetamise vältimine, liigne alkoholitarbimine, liiga palju ja halvasti söömine jne on osteogenesis imperfecta -ga patsientidele rohkem kui diskreetne kasu tervisele, kuna see aeglustab haiguse progresseerumist ja vähendab luude haprust.
SÜMPTOMAATNE RAVI KATSETAMISE ETAPIS
Praegu hindavad arstid ja teadlased mõne sümptomaatilise ravi, sealhulgas kasvuhormoonravi ning bisfosfonaatidel põhineva intravenoosse ja suukaudse ravi efektiivsust.
Hetkel tõotavad eespool nimetatud uurimisravi tulemused head kogu meditsiiniringkonnale.
Prognoos
Osteogenesis imperfecta on negatiivse prognoosiga haigus, kuna see on ravimatu, kahjustab oluliselt elukvaliteeti ja põhjustab mõnel juhul haige subjekti enneaegset surma.
Siiski tuleb märkida, et ka tänu kaasaegsetele sümptomaatilistele ravimeetoditele on paljud kerge osteogeneesi imperfecta vormiga inimesed võimelised elama meeldivat ja rahuldust pakkuvat elu.
Ärahoidmine
Kahjuks puudub praegu ennetav meede osteogenesis imperfecta vastu.